ESMBind (ESMB):ESM-2 用于蛋白质结合位点预测的低秩适应







 此图片来自 Metagenomic Atlas。
此图片来自 Metagenomic Atlas。
简而言之: 将 LoRA 应用于蛋白质语言模型 ESM-2 是一种有效且重要的微调和正则化策略,在我们的训练/测试拆分上,它在蛋白质序列结合位点预测任务中表现出与 SOTA 结构模型相当的性能。然而,由于序列相似性等原因,这可能会产生误导,需要进一步清理数据。该模型仅从单个蛋白质序列预测结合残基,不需要 MSA 或结构信息。继续阅读以更好地了解如何使用 LoRA 微调 pLM,并提供微调您自己的 pLM LoRA 和在您喜欢的蛋白质序列上运行推理的代码示例。
蛋白质语言模型 (pLM),如 ESM-2,是像其大型语言模型 (LLM) 对应物一样的 transformer,但它们是在蛋白质序列而不是自然语言文本上训练的。每个蛋白质序列由 20 种标准氨基酸组成,有时还包括一些非标准氨基酸。通常,对于 ESM-2 模型,每个氨基酸(用一个字母表示)被视为一个 token。因此,一个由 200 个氨基酸组成的蛋白质序列将有 200 个 token 被 pLM 分词。pLM 具有 transformer 的所有常见架构,包括查询、键和值权重矩阵 、 和 ,并且在计算注意力时将每个氨基酸 token 表示为查询、键和值向量。蛋白质语言模型如 ESM-2 使用掩码语言建模目标进行训练,并学习预测被掩码的氨基酸。它们已被证明比 AlphaFold2 更准确地预测蛋白质 3D 结构,并从单个蛋白质序列提供原子级准确和快速的预测。有关我们使用的基本模型的更多信息,请参见此处和此处。
在本文中,我们将讨论应用一种流行的参数高效微调策略,称为低秩适应(Low Rank Adaptation),简称 LoRA。LoRA 在 LLM 社区和 Stable Diffusion 社区中非常流行,但它们也可以用于微调蛋白质语言模型!事实上,它们已被证明作为正则化工具非常有用,并且显著减少了微调 pLM 时出现的过拟合问题,这对于蛋白质来说是一个相当大的障碍,因为存在蛋白质同源物和数据集中高度相似的序列。
什么是...LoRA?
低秩适应(LoRA)是一种参数高效的微调策略,它在 Hugging Face 的 PEFT 库中实现。有关 LoRA 的概念指南,请参见此处。
在深度学习领域,低秩适应(LoRA)的概念最早由 Hu 等人提出。这些 LoRA 为神经网络的传统微调提供了一种高效的替代方案。该过程首先冻结神经网络中现有层的权重。例如,在 Transformer 注意力机制的背景下,这可能涉及冻结查询、键或值矩阵 、 或 的权重。
在此之后,一个 LoRA 层被引入到一个或多个这些预训练权重矩阵中。如果我们认为 是一个冻结的权重矩阵,那么 LoRA 层将采用 的形式,其中 构成了 LoRA。通常,这些是低秩分解,其中 和 ,其中原始权重矩阵为 。通常, 远小于 。
LoRA 的应用只有在 远小于输入和输出维度时才能提供显著的参数减少效益。我们可以选择一个小的 并实现 LoRA 来代替传统的微调。经验证据表明,在许多情况下,选择 或 足以满足需求——即使对于 LLM 中的大型权重矩阵,例如 Transformer 注意力机制的查询、键和值矩阵。在 Stable Diffusion 中,社区训练的 LoRA 的秩通常更高,但目前尚不清楚这是否真的有必要。可能与我们的直觉相反,较低的秩通常更好,特别是对于正则化。
现在让我们探讨一个 LoRA 在减少参数数量方面没有带来任何实质性好处的场景
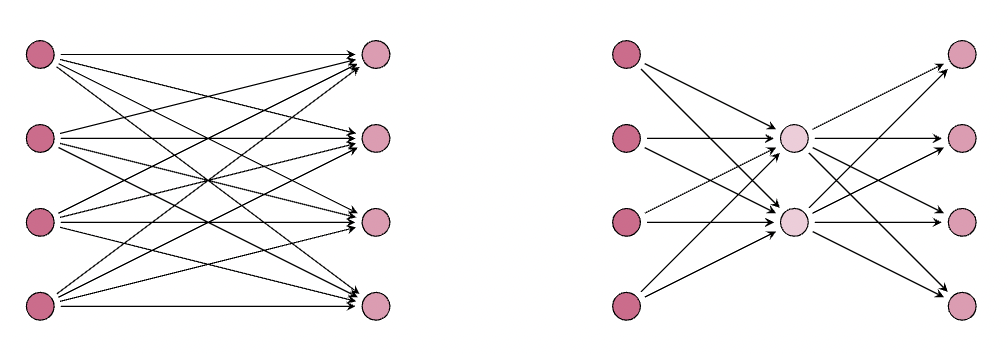
这里,我们看到 LoRA 层 的参数数量与原始层 相同,其中 LoRA(右侧)的参数数量为 ,原始冻结权重矩阵(左侧)的参数数量为 。接下来,让我们看一个例子,它使我们获得冻结权重矩阵参数的 %。
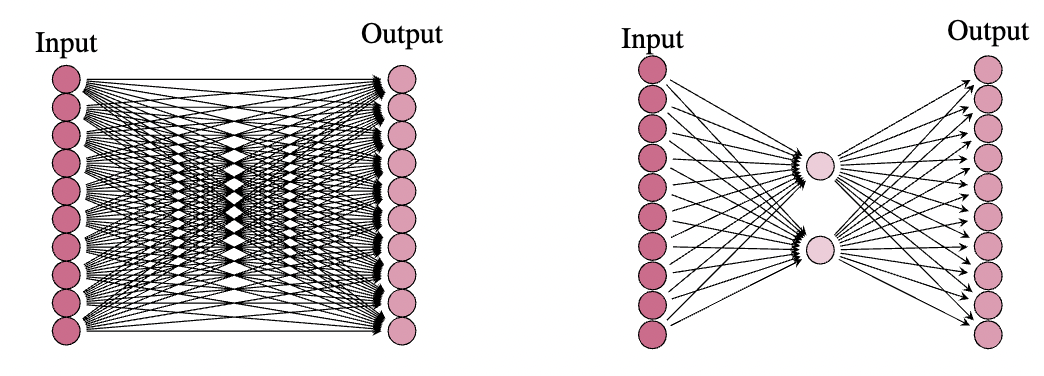
我们看到原始(冻结)权重矩阵有 个参数,而 LoRA 只有 个参数。在大多数情况下,冻结矩阵的秩(这是 LoRA 中间层的神经元数量)远小于输入和输出维度,并且参数数量实际上大幅减少。例如,我们可能有一个输入和输出维度为 ,在这种情况下,权重矩阵有 个参数。然而,这个矩阵的秩通常远低于 。在实践中,结果表明,选择 作为查询、键和值矩阵的中间维度通常足以用于 LoRA。在这种情况下,LoRA 中将有 个参数,这不到原始参数数量的十分之一。一旦我们有了这样的 LoRA,我们就可以在一些下游任务上对其进行训练,然后将 LoRA 权重矩阵 添加到原始(冻结)权重矩阵 中,以获得一个在此新任务上表现良好的模型。
重要的是,LoRA 可以帮助解决过拟合等问题,这在学习蛋白质序列时可能是一个严重的问题。正是这一点,加上参数效率和训练更大模型的需求,促使我们决定采用 LoRA 作为微调策略。此外,使用 Hugging Face PEFT 库进行参数高效微调的 LoRA 的简单性使其成为一个有吸引力的选择。早期也清楚地表明,使用 LoRA 实际上可以提高性能,从而进一步激励我们采用它作为一种策略。
使用 LoRA 进行过拟合和正则化
我们首先对最小的 ESM-2 模型进行了 ~209K 蛋白质序列的普通微调。数据最终根据 UniProt 按家族分类,以帮助解决过拟合和对泛化能力过于乐观的结果,但最初我们没有考虑序列相似性等问题。这可能存在问题,并可能由于训练/测试拆分中存在高度相似的序列而导致过拟合。由于蛋白质同源物等原因,简单随机的数据集拆分不适用于蛋白质。如果数据集未根据序列相似性进行过滤,模型往往会过早过拟合,因为随机训练/测试拆分包含彼此过于相似的序列。
因此,考虑到这一点,我们接下来根据家族划分蛋白质数据,选择随机家族添加到测试集中,直到大约 20% 被分离为测试数据。不幸的是,这对于过拟合的帮助不大。然而,应用 LoRA 确实起到了作用!LoRA 并不能解决所有问题,还需要进一步根据序列相似性过滤数据集,但 LoRA 确实显著减少了过拟合的程度。例如,请参阅 这份 Weights and Biases 报告中提供的示例。此外,请查看使用此策略训练的模型之一 此处。您可能还想查看 这份报告。在阅读模型与 SOTA 结合位点预测模型的比较时,建议谨慎,因为其中一些模型仍然在一定程度上过拟合,并且未在与 SOTA 模型相同的数据集上进行测试。这仅仅是为了粗略了解模型在测试数据集上的表现。
无需 MSA 或结构信息!
由于 ESM-2 和 ESMFold 的架构和训练方式,它们不需要任何**多序列比对(Multiple Sequence Alignment)**。这意味着预测速度更快,所需领域知识更少,使它们更易于使用和访问。这些模型的性能仍然与 AlphaFold2 相当甚至更好,但速度却快了 60 倍!它们也是序列模型,因此不需要蛋白质的任何结构信息。考虑到大多数蛋白质还没有 3D 折叠和骨架结构预测,这是个好消息。这种情况正在缓慢改变,因为 ESMFold 模型提供了快速结构预测,而且 Metagenomic Atlas 现在包含了超过 7.7 亿种蛋白质。尽管这些模型速度快且准确,但它们仍然不如 AlphaFold2 流行,不过人们正在慢慢认识到它们是无价的资源。现在让我们看看一些代码,您可以使用这些代码来训练自己的 ESM-2 模型的 LoRA,以预测蛋白质的结合位点!如果您对深度学习、蛋白质语言模型或蛋白质有所了解,或者即使您不了解,也应该尝试获得更好的指标!此外,如果您熟悉 UniProt 或 UniRef,执行进一步的数据清理可能会有益。
LoRA 推理和训练笔记本供您试用!
为 ESM-2 微调 LoRA
在这里,我们将提供一个如何为 ESM-2 模型微调 LoRA 以预测蛋白质序列结合残基的示例。我们将把这个问题视为一个二元 token 分类任务。开始之前,建议您根据 requirements.txt 文件或 conda-environment.yml 文件设置虚拟环境或 conda 环境。要从 requirements.txt 文件重新创建环境,请使用
pip install -r requirements.txt
要从 conda-environment.yml 文件重新创建 Conda 环境,请使用
conda env create -f conda-environment.yml
导入
import os
# os.environ["CUDA_VISIBLE_DEVICES"] = "0"
import wandb
import numpy as np
import torch
import torch.nn as nn
import pickle
import xml.etree.ElementTree as ET
from datetime import datetime
from sklearn.model_selection import train_test_split
from sklearn.utils.class_weight import compute_class_weight
from sklearn.metrics import (
accuracy_score,
precision_recall_fscore_support,
roc_auc_score,
matthews_corrcoef
)
from transformers import (
AutoModelForTokenClassification,
AutoTokenizer,
DataCollatorForTokenClassification,
TrainingArguments,
Trainer
)
from datasets import Dataset
from accelerate import Accelerator
# Imports specific to the custom peft lora model
from peft import get_peft_config, PeftModel, PeftConfig, get_peft_model, LoraConfig, TaskType
辅助函数和数据预处理
现在,您需要准备训练/测试数据及其标签的 pickle 文件。我们将提供一个关于如何从下载的 UniProt 数据获取自己的 pickle 文件的笔记本,但目前,您可以从这里下载准备好的 pickle 文件。只需导航到“文件和版本”并下载所有四个 pickle 文件到您的机器上。完成此操作后,将以下 pickle 文件路径替换为您下载的 pickle 文件所在的本地路径。我们已将蛋白质序列的截止长度设置为 1000 个氨基酸。这是蛋白质语言模型的“上下文窗口”。请注意,有更小的数据集可用,如果您愿意,可以使用 UniProt 策划自己的数据集。如果您喜欢策划自己的数据,可以尝试在 UniProt 中搜索 (ft_binding:*) 并根据您自己的要求过滤蛋白质。您可能还会考虑从 蛋白质数据库 (PDB) 策划结合位点数据。我们尚未尝试过,但这可能为结合位点提供一个很好的数据来源。
# Helper Functions and Data Preparation
def truncate_labels(labels, max_length):
"""Truncate labels to the specified max_length."""
return [label[:max_length] for label in labels]
def compute_metrics(p):
"""Compute metrics for evaluation."""
predictions, labels = p
predictions = np.argmax(predictions, axis=2)
# Remove padding (-100 labels)
predictions = predictions[labels != -100].flatten()
labels = labels[labels != -100].flatten()
# Compute accuracy
accuracy = accuracy_score(labels, predictions)
# Compute precision, recall, F1 score, and AUC
precision, recall, f1, _ = precision_recall_fscore_support(labels, predictions, average='binary')
auc = roc_auc_score(labels, predictions)
# Compute MCC
mcc = matthews_corrcoef(labels, predictions)
return {'accuracy': accuracy, 'precision': precision, 'recall': recall, 'f1': f1, 'auc': auc, 'mcc': mcc}
def compute_loss(model, inputs):
"""Custom compute_loss function."""
logits = model(**inputs).logits
labels = inputs["labels"]
loss_fct = nn.CrossEntropyLoss(weight=class_weights)
active_loss = inputs["attention_mask"].view(-1) == 1
active_logits = logits.view(-1, model.config.num_labels)
active_labels = torch.where(
active_loss, labels.view(-1), torch.tensor(loss_fct.ignore_index).type_as(labels)
)
loss = loss_fct(active_logits, active_labels)
return loss
# Load the data from pickle files (replace with your local paths)
with open("train_sequences_chunked_by_family.pkl", "rb") as f:
train_sequences = pickle.load(f)
with open("test_sequences_chunked_by_family.pkl", "rb") as f:
test_sequences = pickle.load(f)
with open("train_labels_chunked_by_family.pkl", "rb") as f:
train_labels = pickle.load(f)
with open("test_labels_chunked_by_family.pkl", "rb") as f:
test_labels = pickle.load(f)
# Tokenization
tokenizer = AutoTokenizer.from_pretrained("facebook/esm2_t12_35M_UR50D")
max_sequence_length = 1000
train_tokenized = tokenizer(train_sequences, padding=True, truncation=True, max_length=max_sequence_length, return_tensors="pt", is_split_into_words=False)
test_tokenized = tokenizer(test_sequences, padding=True, truncation=True, max_length=max_sequence_length, return_tensors="pt", is_split_into_words=False)
# Directly truncate the entire list of labels
train_labels = truncate_labels(train_labels, max_sequence_length)
test_labels = truncate_labels(test_labels, max_sequence_length)
train_dataset = Dataset.from_dict({k: v for k, v in train_tokenized.items()}).add_column("labels", train_labels)
test_dataset = Dataset.from_dict({k: v for k, v in test_tokenized.items()}).add_column("labels", test_labels)
# Compute Class Weights
classes = [0, 1]
flat_train_labels = [label for sublist in train_labels for label in sublist]
class_weights = compute_class_weight(class_weight='balanced', classes=classes, y=flat_train_labels)
accelerator = Accelerator()
class_weights = torch.tensor(class_weights, dtype=torch.float32).to(accelerator.device)
自定义加权训练器
接下来,由于我们使用类权重(因为非结合残基和结合残基之间存在不平衡),我们将需要一个自定义加权训练器。
# Define Custom Trainer Class
class WeightedTrainer(Trainer):
def compute_loss(self, model, inputs, return_outputs=False):
outputs = model(**inputs)
loss = compute_loss(model, inputs)
return (loss, outputs) if return_outputs else loss
训练函数
接下来,我们定义训练函数。请注意您可以调整的 LoRA 超参数。尝试调整一些设置,看看能否在数据集上获得更好的性能!关于选择合适的权重矩阵来应用 LoRA,以及选择秩和缩放因子 alpha 等 LoRA 超参数的指南,您可能需要阅读原始论文(本文中已链接)的第 7 节,以及 Weights and Biases 报告(本文中也已链接)。如果您想深入了解选择超参数(尤其是秩)的极度技术细节,您可以训练多个 LoRA 并计算每对 LoRA 权重矩阵的格拉斯曼子空间相似度测量
实现此操作的代码超出了本文的范围,但我们计划在未来的文章中发布如何实现此操作的示例。
def train_function_no_sweeps(train_dataset, test_dataset):
# Set the LoRA config
config = {
"lora_alpha": 1, #try 0.5, 1, 2, ..., 16
"lora_dropout": 0.2,
"lr": 5.701568055793089e-04,
"lr_scheduler_type": "cosine",
"max_grad_norm": 0.5,
"num_train_epochs": 3,
"per_device_train_batch_size": 12,
"r": 2,
"weight_decay": 0.2,
# Add other hyperparameters as needed
}
# The base model you will train a LoRA on top of
model_checkpoint = "facebook/esm2_t12_35M_UR50D"
# Define labels and model
id2label = {0: "No binding site", 1: "Binding site"}
label2id = {v: k for k, v in id2label.items()}
model = AutoModelForTokenClassification.from_pretrained(model_checkpoint, num_labels=len(id2label), id2label=id2label, label2id=label2id)
# Convert the model into a PeftModel
peft_config = LoraConfig(
task_type=TaskType.TOKEN_CLS,
inference_mode=False,
r=config["r"],
lora_alpha=config["lora_alpha"],
target_modules=["query", "key", "value"], # also try "dense_h_to_4h" and "dense_4h_to_h"
lora_dropout=config["lora_dropout"],
bias="none" # or "all" or "lora_only"
)
model = get_peft_model(model, peft_config)
# Use the accelerator
model = accelerator.prepare(model)
train_dataset = accelerator.prepare(train_dataset)
test_dataset = accelerator.prepare(test_dataset)
timestamp = datetime.now().strftime('%Y-%m-%d_%H-%M-%S')
# Training setup
training_args = TrainingArguments(
output_dir=f"esm2_t12_35M-lora-binding-sites_{timestamp}",
learning_rate=config["lr"],
lr_scheduler_type=config["lr_scheduler_type"],
gradient_accumulation_steps=1,
max_grad_norm=config["max_grad_norm"],
per_device_train_batch_size=config["per_device_train_batch_size"],
per_device_eval_batch_size=config["per_device_train_batch_size"],
num_train_epochs=config["num_train_epochs"],
weight_decay=config["weight_decay"],
evaluation_strategy="epoch",
save_strategy="epoch",
load_best_model_at_end=True,
metric_for_best_model="f1",
greater_is_better=True,
push_to_hub=False,
logging_dir=None,
logging_first_step=False,
logging_steps=200,
save_total_limit=7,
no_cuda=False,
seed=8893,
fp16=True,
report_to='wandb'
)
# Initialize Trainer
trainer = WeightedTrainer(
model=model,
args=training_args,
train_dataset=train_dataset,
eval_dataset=test_dataset,
tokenizer=tokenizer,
data_collator=DataCollatorForTokenClassification(tokenizer=tokenizer),
compute_metrics=compute_metrics
)
# Train and Save Model
trainer.train()
save_path = os.path.join("lora_binding_sites", f"best_model_esm2_t12_35M_lora_{timestamp}")
trainer.save_model(save_path)
tokenizer.save_pretrained(save_path)
训练!
运行以下命令开始训练您的 LoRA!请注意,由于数据集大小的原因,这可能需要一段时间,具体取决于您的 GPU。如果您想在 Colab 中运行此操作,您可能需要使用 Colab Pro,或者训练一个较小的模型和/或使用一个较小的数据集。但是,运行推理(见下文)可以在标准 Colab 中完成。
train_function_no_sweeps(train_dataset, test_dataset)
检查训练/测试指标
最后,您可以通过将以下代码中的 LoRA 模型路径 AmelieSchreiber/esm2_t12_35M_lora_binding_sites_v2_cp3 替换为您训练的 LoRA 检查点路径来检查其中一个保存模型的训练/测试指标。这将帮助您检查过拟合以及模型对未见蛋白质序列的泛化能力。您的训练/测试指标应该彼此相似。也就是说,您的训练指标应该与您的测试指标大致相同。如果训练指标比测试指标差,您可能需要训练更长时间,因为模型可能欠拟合。如果您的训练指标远高于您的测试指标,则您的模型已过拟合!
from sklearn.metrics import(
matthews_corrcoef,
accuracy_score,
precision_recall_fscore_support,
roc_auc_score
)
from peft import PeftModel
from transformers import DataCollatorForTokenClassification
# Define paths to the LoRA and base models
base_model_path = "facebook/esm2_t12_35M_UR50D"
lora_model_path = "AmelieSchreiber/esm2_t12_35M_lora_binding_sites_v2_cp3" # "path/to/your/lora/model" Replace with the correct path to your LoRA model
# Load the base model
base_model = AutoModelForTokenClassification.from_pretrained(base_model_path)
# Load the LoRA model
model = PeftModel.from_pretrained(base_model, lora_model_path)
model = accelerator.prepare(model) # Prepare the model using the accelerator
# Define label mappings
id2label = {0: "No binding site", 1: "Binding site"}
label2id = {v: k for k, v in id2label.items()}
# Create a data collator
data_collator = DataCollatorForTokenClassification(tokenizer)
# Define a function to compute the metrics
def compute_metrics(dataset):
# Get the predictions using the trained model
trainer = Trainer(model=model, data_collator=data_collator)
predictions, labels, _ = trainer.predict(test_dataset=dataset)
# Remove padding and special tokens
mask = labels != -100
true_labels = labels[mask].flatten()
flat_predictions = np.argmax(predictions, axis=2)[mask].flatten().tolist()
# Compute the metrics
accuracy = accuracy_score(true_labels, flat_predictions)
precision, recall, f1, _ = precision_recall_fscore_support(true_labels, flat_predictions, average='binary')
auc = roc_auc_score(true_labels, flat_predictions)
mcc = matthews_corrcoef(true_labels, flat_predictions) # Compute the MCC
return {"accuracy": accuracy, "precision": precision, "recall": recall, "f1": f1, "auc": auc, "mcc": mcc} # Include the MCC in the returned dictionary
# Get the metrics for the training and test datasets
train_metrics = compute_metrics(train_dataset)
test_metrics = compute_metrics(test_dataset)
train_metrics, test_metrics
运行推理
现在,您已经有了一个可以预测结合位点的训练好的 LoRA。您可能希望在您喜欢的蛋白质序列上运行推理。为此,只需运行以下代码(用您自己的模型替换下面的模型)。如果您只想测试 Hugging Face 上已有的微调模型,您可以独立于上面的其余代码运行此操作,而无需进行任何更改。
!pip install transformers -q
!pip install peft -q
from transformers import AutoModelForTokenClassification, AutoTokenizer
from peft import PeftModel
import torch
# Path to the saved LoRA model
model_path = "AmelieSchreiber/esm2_t12_35M_lora_binding_sites_v2_cp3"
# ESM2 base model
base_model_path = "facebook/esm2_t12_35M_UR50D"
# Load the model
base_model = AutoModelForTokenClassification.from_pretrained(base_model_path)
loaded_model = PeftModel.from_pretrained(base_model, model_path)
# Ensure the model is in evaluation mode
loaded_model.eval()
# Load the tokenizer
loaded_tokenizer = AutoTokenizer.from_pretrained(base_model_path)
# Protein sequence for inference
protein_sequence = "MAVPETRPNHTIYINNLNEKIKKDELKKSLHAIFSRFGQILDILVSRSLKMRGQAFVIFKEVSSATNALRSMQGFPFYDKPMRIQYAKTDSDIIAKMKGT" # Replace with your actual sequence
# Tokenize the sequence
inputs = loaded_tokenizer(protein_sequence, return_tensors="pt", truncation=True, max_length=1024, padding='max_length')
# Run the model
with torch.no_grad():
logits = loaded_model(**inputs).logits
# Get predictions
tokens = loaded_tokenizer.convert_ids_to_tokens(inputs["input_ids"][0]) # Convert input ids back to tokens
predictions = torch.argmax(logits, dim=2)
# Define labels
id2label = {
0: "No binding site",
1: "Binding site"
}
# Print the predicted labels for each token
for token, prediction in zip(tokens, predictions[0].numpy()):
if token not in ['<pad>', '<cls>', '<eos>']:
print((token, id2label[prediction]))
后续步骤
以类似于 Chinchilla 论文的方式,以 1:1 的方式扩展模型和数据集,已显示出性能提升,尽管过拟合的障碍尚未在所有数据集上完全解决。关于蛋白质语言模型是否与 LLM 遵循相似的缩放定律,目前 OpenBioML 社区正在积极研究,如果您对这篇文章感兴趣,欢迎加入!该项目的下一步将是根据序列相似性进一步过滤数据集,以进一步减轻过拟合并提高泛化能力。我们发现 LoRA 显著改善了过拟合问题,这令人着迷,并计划继续尝试应用该技术。
我们还计划使用量化低秩适应(QLoRA)来帮助扩展到更大的模型。然而,在撰写本文时,Hugging Face 的 ESM-2 模型移植版本尚不支持梯度检查点。如果您想改变这一点,请向 Hugging Face Transformers Github 提交拉取请求,以便我们能够为 Hugging Face 的 ESM-2 模型移植版本启用梯度检查点!由于 LoRA 和缩放所带来的迄今为止令人鼓舞的改进,我们希望仅基于序列的方法能够达到与 SOTA 相当的性能。这将是一个有价值的贡献,因为大多数蛋白质尚未进行 3D 折叠和骨架结构预测。我们还希望这种简单而有效的微调策略能够降低那些希望涉足使用和微调蛋白质语言模型的人的进入门槛,并使 ESM-2 模型的全部潜力得到更好的实现。在未来的工作中,我们还计划研究诸如翻译后修饰(PTM)预测(将其视为 token 分类任务),以及蛋白质功能预测任务(如 CAFA-5),也使用 LoRA。我们已经在为其中一些任务准备了笔记本,供您尝试更多的 LoRA 微调!





