蛋白质相似度和套娃式嵌入



背景
2022年,UniProt为数据库中的每个经过验证的蛋白质添加了嵌入。这些嵌入代表构成该蛋白质的氨基酸序列,可用于测量相似性和构建搜索应用程序。
最近,机器学习领域对重新思考嵌入以使向量数据库更快、更高效产生了兴趣。在阅读了套娃式嵌入后,我提出了一个训练适应性蛋白质模型的项目。概括来说,其概念是让嵌入中较早的索引承载更多的意义。您可以使用较短的嵌入(而不是UniProt使用的完整1,024维)来获得近似结果。此领域中的项目通常会先使用较短的嵌入进行搜索,然后再根据完整的嵌入进行重新排序——Supabase 博客文章。
嵌入数据
我需要使用从UniProt当前嵌入中获得的相似性分数来训练蛋白质对模型。
如何选择合适的训练对?我考虑了两个因素
- 我需要在单独的训练和测试组中进行配对。我决定使用来自khairi/uniprot-swissprot数据集的训练/测试/验证拆分。
- 配对应该代表较小和较大差异的混合(即不仅仅是“这些蛋白质看起来一点都不相似”)。
让我们看看10万个随机蛋白质对,并计算余弦距离。我曾担心这些蛋白质会聚集在某个地方,但这却是一个有趣的分布,平均值为0.674。
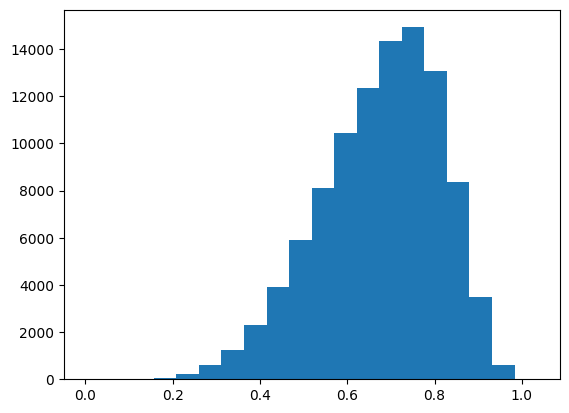
我想对于每种蛋白质,我应该随机配对两个,然后找到距离小于0.55和大于0.8的对(大约是随机配对距离的前五分位数和后五分位数)。
蛋白质对和距离数据集:protein-pairs-uniprot-swissprot
CoLab Notebook:https://colab.research.google.com/drive/1rhoF0pAauHbiaNHdZkOI5O7A3mMcvS6G?usp=sharing
agemagician/uniref50数据集将是蛋白质对的替代来源,但它包含未经审查/未经验证的蛋白质,UniProt未为其预计算嵌入。
训练
UniProt 使用 Rostlab/prot_t5_xl_uniref50 模型生成其嵌入。对于我的基础模型,我使用同一实验室的较小 BERT 模型 prot_bert_bfd。
分词器基于 IUPAC-IUB 代码,其中每个氨基酸映射到一个字母。注意:文本输入中每个字母之间必须有空格(例如“M S L E Q”)。
好消息是这个 BERT 基础模型已经预训练了氨基酸序列,我只需要在模型周围使用 SentenceTransformers 包装器来使用 Matryoshka 损失进行训练。
在 V100 GPU 上,当批处理大小为 2 时运行顺利后,我将批处理大小增加到 10 并在 A100 上运行。我运行了大约 3.5 小时;对于完整的训练集,这将需要超过 20 小时。
嵌入模型:https://huggingface.co/monsoon-nlp/protein-matryoshka-embeddings
CoLab Notebook:https://colab.research.google.com/drive/1uBk-jHOAPhIiUPPunfK7bMC8GnzpwmBy
评估
在训练过程中,我使用了 SentenceTransformers 的 EmbeddingSimilarityEvaluator 来检查完整的嵌入是否在验证集上有效并有所改进。
| 步骤 | 余弦皮尔逊相关系数 | 余弦斯皮尔曼相关系数 |
|---|---|---|
| 3000 | 0.8598688660086558 | 0.8666855900999677 |
| 6000 | 0.8692703523988448 | 0.8615673651584274 |
| 9000 | 0.8779733537629968 | 0.8754158959780602 |
| 12000 | 0.8877422045031667 | 0.8881492475969834 |
| 15000 | 0.9027359688395733 | 0.899106724739699 |
| 18000 | 0.9046675789738002 | 0.9044183600191271 |
| 21000 | 0.9165801536390973 | 0.9061381997421003 |
| 24000 | 0.9128046401341833 | 0.9076748537082228 |
| 27000 | 0.918547416546341 | 0.9127677526055185 |
| 30000 | 0.9239429677657788 | 0.9187051589781693 |
我们仍然需要验证这些嵌入即使在缩短后也仍然有用。
我从测试集中随机选取了 2,000 对蛋白质,并比较了原始模型 (x) 和新的套娃式模型 (y) 的余弦距离。关于此图有一些需要思考的地方,但最主要的结论是它们在测试集上具有相关性。
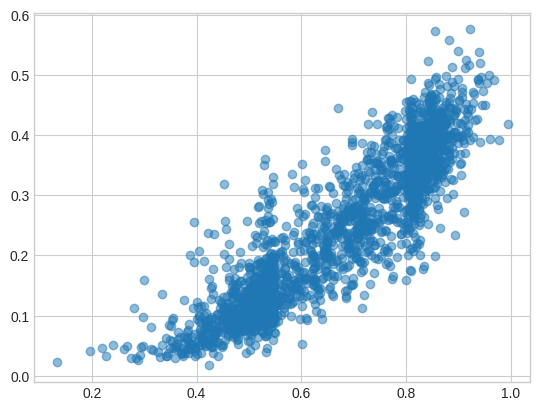
128 维嵌入(不同的随机对)的图看起来非常相似!
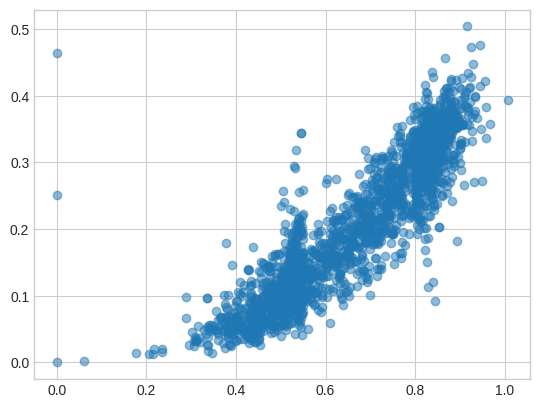
如果我使用原始模型的前 128 维,它们虽然与完整尺寸的嵌入保持接近,但仍有明显的漂移。
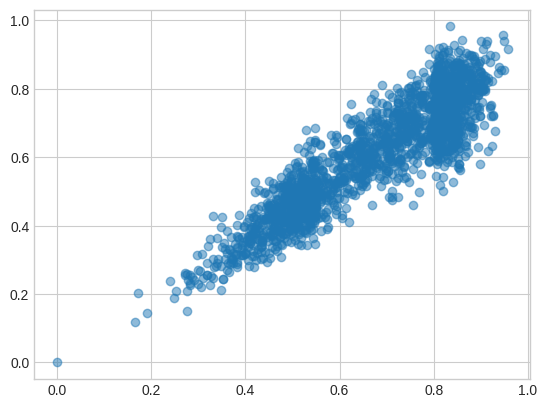
CoLab Notebook:https://colab.research.google.com/drive/1hm4IIMXaLt_7QYRNvkiXl5BqmsHdC1Ue?usp=sharing
🦠🧬🤖🪆 未来思考
想在生物LLM方面合作吗?联系我们!。
我想做一个关于植物 DNA 的项目,并可能与作物基因组学研究(土豆、藜麦、小麦、水稻等)建立联系。
这些嵌入是否足够有趣,值得联系 UniProt 团队?或者获得资源来在整个数据集上运行 T5 模型?
套娃式嵌入论文也对较小嵌入在分类任务上的表现进行了测试。看起来我可以使用这段代码中的 MRL_Linear_Layer 来改变嵌入大小。我目前的想法是在TAPE的任务上进行测试。
在我研究这个的同时,HuggingFace、Cohere 和 pgvecto.rs 发布了关于量化嵌入的博客文章?或许这也会有所帮助。